НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ НЕРВНОЙ СИСТЕМЫ
ЛЕКЦИЯ 16
Дегенеративные заболевания с преимущественным поражением нервно-мышечного аппарата составляют самую значительную группу среди всех наследственных заболеваний.
Исключительно важными, а нередко решающими в диагностике нервно-мышечных заболеваний являются результаты электрофизиологических и биохимических исследований. Столь же велико значение патоморфологических находок. Изучение мышечного биоптата в световом микроскопе помогает дифференцировать миогенную атрофию от неврогенной. Гистохимическое исследование необходимо для выявления метаболических поражений мышц, а электронная микроскопия открыла целый большой класс заболеваний - непрогрессирующие миопатии.
Прогрессирующие мышечные дистрофии. Термином мышечные дистрофии называется группа генетически детерминированных расстройств, характеризующихся прогрессирующими дегенеративными изменениями в мышечных волокнах без первичной патологии периферического (нижнего) мотонейрона.
Различные формы отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных атрофии, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.
Большинство мышечных дистрофий хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросыих патогенеза и лечения остаются до сего времени неразрешенными. Большие надежды возлагаются на молекулярную генетику, с помощью которой определено местонахождение генов уже многих нозологических форм.
Диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число членов семьи затрудняет определение типа наследования.
Характерным моторным дефектом у больных с мышечными дистрофиями является “утиная” походка: больной ходит переваливаясь к боку на бок. Она связана главным образом со слабостью ягодичных мышц, прежде всего средней и малой, которые фиксируют таз относительно бедренной кости. В результате при заболевании возникает наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). При ходьбе сторона наклона постоянно меняется. Указанные изменения можно проверить и в пробе Тренделенбурга, попросив больного поднять одну ногу, согнув ее под прямым углом в коленном и тазобедренном суставах: таз на стороне поднятой ноги опускается (а не поднимаетсякак в норме) из-за слабости средней ягодичной мышцы опорной ноги.
Поднимаясь из горизонтального положения, больной с выраженной мышечной слабостью проксимальных мышц с трудом переворачивается на живот, затем, упираясь руками в пол, становится на четвереньки и после этого, упираясь руками в голени, затем в бедра, постепенно выпрямляется. Этот феномен “набирания по себе” носит название приема Говерса. Часто он связан со слабостью больших ягодичных мышц.
Миодистрофия Дюшенна. Псевдогипертрофическая мышечная дистрофия Дюшенна встречается чаще всех других заболеваний мышечной системы (30 на 100000 живых новорожденных). Характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У детей более раннего возраста начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать. Больные умирают на 2-3-м десятилетии жизни.
Одними из первых признаков заболевания являются уплотнения икроножных мышц и постепенное увеличениеихобъема за счет псевдогипертрофий. Атрофии мышц бедра, тазового пояса нередко маскируются хорошо развитой подкожной жировой клетчаткой. Постепенно процесс принимает восходящее направление и распространяется за плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук.
В терминальной стадии слабость мышц может распространяться на мышцы лица, глотки, дыхательные мышцы.
В развитой стадии болезни имеются такие характерные симптомы, как “утиная походка”; подчеркнутый поясничный лордоз, крыловидные лопатки, симптом “свободных надплечий”. Типичны ранние мышечные контрактуры и сухожильные ретракции, особенно ахилловых сухожилий. Рано выпадают коленные рефлексы, а затем рефлексы с верхних конечностей.
Псевдогипертрофии могут развиваться не только в икроножных, но также в ягодичных, дельтовидных мышцах, мышцах живота, языка. Очень часто страдает сердечная мышца по типу кардиомиопатии. Выявляются нарушения ритма сердечной деятельности, расширение границ сердца, глухость тонов, изменения ЭКГ. Острая сердечная недостаточность - наиболее частая причина летальных исходов при миодистрофии Дюшенна. На вскрытии находят фиброз и жировую инфильтрацию сердечной мышцы.
Нередко наблюдается нарушение моторики желудочно-кишечного тракта.
Частым симптомом является снижение интеллекта. Представляет интерес тот факт, что в одних семьях олигофрения бывает резко выражена, в других сравнительно умеренно. Изменение высших психических функций обычно не прогрессирует и не коррелирует с тяжестью мышечного дефекта. Оно не может быть объяснено только педагогической запущенностью больных детей, которые рано выключаются из детских коллективов, не посещают детский сад и школу из-за двигательных дефектов. При КТ и МРТ нередко обнаруживают церебральную атрофию, возможно, связанную с нарушением пренатального развития головного мозга.
Нередко у детей развивается адипозогенитальный синдром, иногда и другие признаки эндокринной недостаточности. Часто находят изменения в костной системе: деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз.
Отличительной особенностью формы Дюшенна является высокая степень гиперферментемии уже на ранних стадиях развития процесса. Так, уровень специфического для мышечной ткани фермента - креатининфосфокиназы - в сыворотке крови может превышать в десятки и даже сотни раз нормальные показатели. Резкое (в 10-100 раз) увеличение креатининфосфокиназы (КФК) при нервно-мышечной патологии должно побуждать к обсуждению прежде всего следующих заболеваний: болезни Дюшенна, болезни Беккера, полиомиозита и дерматомиозита, пароксизмалыюй миоглобулинурии, дистальной миодистрофии. Лишь в далеко зашедших стадиях болезни степень гиперферментемии постепенно снижается. Имеются сообщения о повышении КФК на стадии внутриутробного развития.
Миодистрофия Дюшенна передается по рецессивному, сцепленному с Х-хромосомой типу. Ген локализован в коротком плече Х-хромосомы. Довольно высока частота мутации гена (30%), что объясняет большое количество спорадических случаев.
Мутация (чаще всего делеция) приводит к половому или почти полному отсутствию продукта гена - структурного белка дистрофика. Физиологическая роль дистрофика до конца не установлена. Он обнаруживается в больших концентрациях в области сарколеммы, играя, видимо, определенную роль в поддержании целости этой мембраны. Отсутствие дистрофика вызывает структурные изменения в сарколемме, что в свою очередь приводит к потере внутриклеточных компонентов и повышенному входу кальция, что в конечном счете ведет к гибели миофибрилл. Полагают, что дефицит дистрофика в синаптических зонах корковых нейронов является причиной задержки психического развития.
Для медико-генетического консультирования очень важно установление гетерозиготного носительства. При миодистрофии Дюшенна у гетерозигот приблизительно в 70% случаев выявляются субклинические, а иногда и явные признаки мышечной патологии - некоторое уплотнение и даже увеличение икроножных мышц, быстрая утомляемость мышц при физической нагрузке, изменения на ЭМГ и при патоморфологическом исследовании мышечных биоптатов. Наиболее часто у гетерозиготных носительниц выявляется увеличение активности креатининфосфокиназы.
При наличии клинической картины миодистрофии Дюшенна у лиц женского пола следует в первую очередь исключить возможность аномалии по Х-хромосоме - синдром Шерешевского-Тернера (ХО), синдром Морриса (XY) или мозаицизм по этим синдромам.
Мышечная дистрофия Дюшенна, начинающая развиваться еще в пренатальном периоде, является по сути врожденной миопатией и может быть диагностирована вскоре после рождения путем проведения мышечной биопсии и определения активности КФК.
Миодистрофия Беккера. Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофией Дюшенна существует доброкачественная форма - болезнь Беккера. По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягко, больные длительно сохраняют работоспособность, в возрасте 20- 30 лет и позже еще могут ходить. Фертильность не снижена, поэтому заболевание иногда прослеживается в нескольких поколениях семьи: больной мужчина через свою дочь передает заболевание внуку (“эффект деда”). Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракция пяточных (ахилловых) сухожилий выражена менее резко, чем при болезни Дюшенна.
При этой форме не отмечается нарушений интеллекта, кардиомиопатия отсутствует или выражена незначительно.
Как и при других Х-сцепленных миодистрофиях, при форме Беккера значительно повышается активность КФК, хотя и в меньшей степени, чем при болезни Дюшенна, не превышая 5000 ед. Ген болезни Беккера, как и болезни Дюшенна, локализуется в коротком плече Х-хромосомы; вероятно, оба локуса тесно связаны между собой или являются аллельными. В отличие от болезни Дюшенна, при которой дистрофии практически отсутствует, при болезни Беккера синтезируется аномальный дистрофии. Отличия обнаруживаются и при мышечной биопсии. При миодистрофии Беккера мышечные волокна обычно неокруглы, гиалиновые волокна, характерные для миодистрофии Дюшенна, наблюдаются крайне редко.
Миодистрофия Ландузи-Дежерина (лицелопаточно-плечевая миодистрофия). Заболевание передается по аутосомно-доминантному типу с высокой пенетрантностью, но несколько вариабельной экспрессивностью. Встречается гораздо реже, чем миодистрофия Дюшенна (0,4 на 100 тыс. населения). Предполагают, что ген этого заболевания локализован в 4-й хромосоме. Женщины болеют чаще мужчин (3:1), Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.
Миодистрофия Ландузи-Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она еще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике, удается выявить некоторую слабость мышц, например мышц лица, и в более раннем возрасте.
Мышечная слабость и атрофия вначале появляются в мышцах лица или плечевого пояса. Постепенно эти нарушения распространяются на мышцы проксимальных отделов рук, а затеми на нижние конечности. В большинстве случаев вначале поражаются мышцы передней поверхности голеней (с развитием свисающей стопы), затем мышцы проксимальных отделов ног. На высоте заболевания грубо страдают круговые мышцы глаза и рта, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая, трехглавая мышцы плеча. Характерен внешний вид больных: типичное лицо миопата с “поперечной улыбкой” (“улыбка Джоконды”), протрузией верхней губы (“губы тапира”), резко выраженные крыловидные лопатки, своеобразная деформация грудной клетки с уплощением ее в переднезаднем направлении и ротацией внутрь плечевых суставов. Нередко имеется асимметрия поражения, даже в пределах одной мышцы (например, круговой мышцы рта). Может наблюдаться псевдогипертрофия икроножных, дельтовидных мышц, иногда мышц лица. Контрактуры и ретракции выражены умеренно. Сухожильные рефлексы длительное время бывают сохранены, но иногда снижаются уже на ранней стадии.
Признаки поражения сердечной мышцы выявляются редко. Активность сывороточных ферментов увеличена незначительно и может быть нормальной. Интеллект не страдает. Продолжительность жизни в большинстве случаев не снижается. Представляет интерес тот факт, что ЭМГ при миодистрофии Ландузи-Дежерина нередко не совсем типична для мышечного уровня поражения. У некоторых больных (членов одной семьи) может наблюдаться снижение амплитуды биопотенциалов, интерференционный тип кривой, у других, наоборот, уменьшение частоты и гиперсинхронная активность, иногда с типичным ритмом частокола. Следует помнить о спинальном варианте, имитирующем болезнь Ландузи-Дежерина.
Миодистрофия Эрба-Рота (конечностно-поясная миодистрофия). Передается по аутосомно-рецессивному типу, оба пола страдают одинаково. Начало заболевания в большинстве случаев относится к середине 2-го десятилетия жизни (14-16 лет), однако описана как ранняя, псевдодюшенновская форма, когда первые симптомы проявляются в возрасте до 10 лет и заболевание протекает тяжело, так и поздний вариант с началом после 30 лет.
Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидизация наступает через 15-20 лет от начала появления первых симптомов. Миодистрофия начинается либо с поражения мышц тазового пояса и проксимальных отделов ног (форма Лейдена-Мебиуса), либо с плечевого пояса (форма Эрба). В некоторых случаях плечевой и тазовый пояса поражаются одновременно. Довольно значительно страдают мышцы спины и живота. У больных имеется характерная “утиная” походка, затруднено вставание из положения лежа и сидя, подчеркнут поясничный лордоз. Мышцы лица в большинт стве случаев не страдают. Для этой формы малохарактерны контрактуры и псевдогипертрофии. Могут иметь место концевые атрофии и сухожильные ретракции. Интеллект обычно сохранен. Сердечная мышца большей частью не поражена. Уровень ферментов в сыворотке крови, как правило, повышен, однако не столь резко, как при Х-сцепленной миодистрофии. Есть указания, что у больных мужского пола уровень КФК выше, чем у больных женщин. Имеется значительная разница в экспрессивности мутантного гена у разных членов семьи - наряду с тяжелой клинической картиной могут быть сравнительно легкие и даже стертые клинические симптомы. Смерть обычно наступает от легочных осложнений.
Поскольку клинику конечностно-поясной миодистрофии особенно охотно имитируют нервно-мышечные заболевания иного характера, необходимо, особенно в спорадических случаях и при позднем начале заболевания, проводить тщательное клиническое обследование для исключения спинальной амиотрофии, полимиозита, метаболических, эндокринных, токсических, лекарственных, карциноматозных миопатий. В прошлом имела место явная гипердиагностика этой формы мышечных дистрофий.
Лечение мышечных дистрофий. Терапевтические возможности при мышечных дистрофиях весьма ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и, возможно, на некоторое снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Лечебный комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию.
Лечебная гимнастика состоит из пассивных и активных движений, выполняемых во всех суставах в различных положениях: стоя, сидя, лежа, при различном положении конечностей. Активные движения предпочтительнее выполнять в изометрическом режиме. Занятия гимнастикой необходимо проводить регулярно по нескольку раз в день. В то же время следует предостеречь от чрезмерных упражнений, особенно сопровождающихся перерастяжением мышц. Важное значение (особенно после иммобилизации больного) имеют дыхательные упражнения.
Ортопедические мероприятия консервативного (специальные шины) и оперативного характера (ахиллотомия, пересечение икроножной мышцы), направленные на коррекцию контрактур и формирующихся патологических установок конечностей, также имеют целью сохранить возможность самостоятельного передвижения. При этом в каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. Следует учитывать, что нередко (в частности, при выраженном гиперлордозе и слабости четырехглавой мышцы бедра) эквиноварусная установка стоп имеет компенсаторное значение и после проведения, например, ахиллотомии больной может оказаться окончательно обездвиженным. При развивающихся контрактурах рекомендуется проводить осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Медикаментозная терапия предполагает назначение препаратов метаболического действия, направленных на восполнение энергетического и белкового дефицита, однако их эффективность весьма сомнительна. Применяют антагонисты кальция (в связи с выявленным при болезни Дюшенна дефектом клеточных мембран, приводящим к повышенному поступлению кальция внутрь клетки), иммуномодуляторы, фосфорсодержащие соединения (АТФ, фосфаден), витамин Е (100мг внутрь 3 раза в день). Показано, что при болезни Дюшенна применение преднизолона (0,75 мг/кг в сутки) может драматически увеличивать силу мышц, однако этот эффект сохраняется неболее года и в целом не влияет на исход заболевания. В связи с серьезными побочными эффектами, возникающими ври длительном применении препарата, его использование нецелесообразно. Оценки эффекта анаболических стероидов противоречивы и их назначение зачастую сопряжено с неоправданным риском. Оценивая эффект тех или иных препаратов при болезни Дюшенна, следует учитывать, что при умеренной выраженности заболевания у больных в возрасте 3-6 лет может отмечаться относительная стабилизация состояния, связанная с возрастным развитием мышечной системы, приобретением двигательных навыков, что может в какой-то степени временно скомпенсировать непрерывно текущий дистрофический процесс.
Определенное значение имеет коррекция питания больного, рекомендуется диета с высоким содержанием белка и низким содержанием жиров и пониженной калорийностью при оптимальном содержании витаминов и микроэлементов. Важную роль играет психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
Нервно-мышечные заболевания (НМЗ) - это наиболее многочисленная группа наследственных заболеваний, в основе которых лежит генетически детерминированное поражение передних рогов спинного мозга, периферических нервов и скелетных мышц.
К нервно-мышечным заболеваниям относятся:
1) прогрессирующие мышечные дистрофии (первичные миопатии);
2) спинальные и невральные амиотрофии (вторичные миопатии);
3) врожденные непрогрессирующие миопатии;
4) нервно-мышечные заболевания с миотоническим синдромом;
5) пароксизмальные миоплегии;
6) миастения.
15.2. Прогрессирующие мышечные дистрофии (первичные миопатии)
Прогрессирующие мышечные дистрофии (ПМД), или первичные миопатии, характеризуются дегенеративными изменениями в мышечной ткани.
Патоморфологические изменения при ПМД характеризуются истончением мышц, заменой их жировой и соединительной тканью. В саркоплазме выявляются очаги фокального некроза, ядра мышечных волокон располагаются цепочками, мышечные волокна теряют поперечную исчерченность.
Вопросы патогенеза остаются до настоящего времени неразрешенными. В основе миопатии лежит дефект мембраны мышечных клеток. Большие надежды возлагаются на молекулярную генетику.
Различные формы миопатии отличаются типом наследования, сроками начала процесса, характером и быстротой его течения и топографией мышечных атрофии.
Миопатии клинически характеризуются слабостью и атрофией мышц. Существуют различные формы ПМД.
15.2.1. Миодистрофия Дюшенна (псевдогипертрофическая форма пмд)
Встречается наиболее часто из всех ПМД (30:100 000). Данная форма характеризуется ранним началом (2-5 лет) и злокачественным течением, болеют преимущественно мальчики. Миопатия Дюшенна наследуется по рецессивному типу, сцепленному с Х-хромосомой. Патологический ген локализуется в коротком плече хромосомы (X, или 21-й хромосомы).
Довольно высока мутация гена, чем объясняется значительная частота спорадических случаев. Мутация (чаще всего делеция) гена приводит к отсутствию дистрофина в мембране мышечных клеток, что приводит к структурным изменениям сарколеммы. Это способствует выходу кальция и ведет к гибели миофибрилл.
Одним из первых признаков заболевания является уплотнение икроножных мышц и постепенное увеличение их объема за счет псевдогипертрофий. Процесс носит восходящий характер. Для развернутой стадии заболевания характерна «утиная» походка, больной ходит, переваливаясь с боку на бок, что связано главным образом со слабостью ягодичных мышц.
В результате этого происходит наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). При ходьбе сторона наклона все время меняется. Это можно проверить в позе Тренделенбурга, попросив больного поднять одну ногу, согнув ее под прямым углом в коленном и тазобедренном суставе: таз на стороне поднятой ноги опускается (а не поднимается как в норме) из-за слабости средней ягодичной мышцы опорной ноги.
При миопатии Дюшенна часто отмечается выраженный лордоз, крыловидные лопатки, типичные мышечные контрактуры, рано выпадают коленные рефлексы. Нередко удается обнаружить изменения в костной системе (деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз). Может отмечаться снижение интеллекта и различные эндокринные расстройства (адипозогенитальный синдром, синдром Иценко-Кушинга). К 14-15 годам больные обычно уже полностью обездвижены, в терминальной стадии слабость может распространиться на мышцы лица, глотки, диафрагмы. Погибают они чаще всего на 3-м десятилетии жизни от кардиомиопатии или присоединения интеркуррентных инфекций.
Отличительной особенностью миопатии Дюшенна является резкое повышение специфического мышечного фермента - креатинфосфокиназы (КФК) в десятки и сотни раз, а также повышение миоглобина в 6-8 раз.
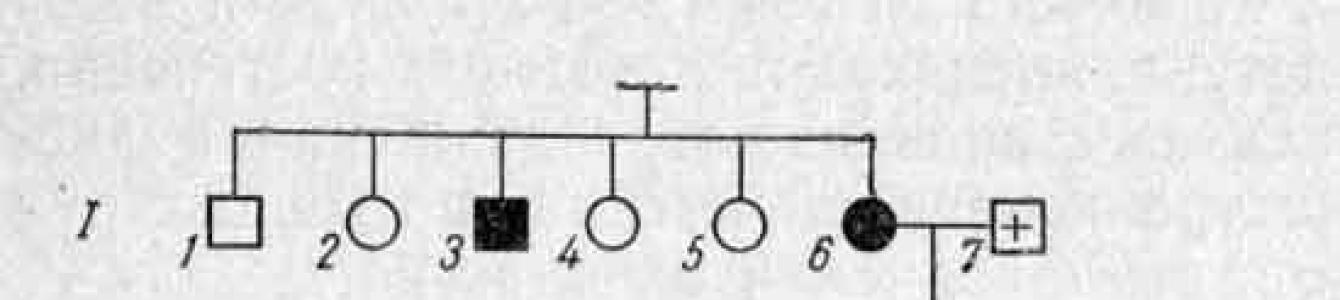
Для медико-генетического консультирования важно установление гетерозиготного носительства. У 70% гетерозигот определяются субклинические и клинические признаки мышечной патологии: уплотнение и увеличение икроножных мышц, быстрая утомляемость мышц при физической нагрузке, изменение мышечных биоптатов и биопотенциалов по данным ЭМГ.
Наследственно-дегенеративные заболевания нервной системы - это большая группа болезней, обусловленная генетической патологией. Врожденные болезни не всегда являются наследственными. Наиболее частыми являются: 1) наследственные нервно-мышечные заболевания,
2) наследственные болезни обмена, протекают с поражением нервной системы, 3) факоматозы и системные дегенерации
Наследственные болезни нервно-мышечной системы
Большая часть детских заболеваний является наследственной. Они характеризуются поражением мышечной ткани, периферических нейронов и нередко спинного мозга. Проявляются в виде мышечной слабости, повышенной утомляемости, низкого мышечного тонуса с последующим развитием мышечной дистрофии. Диагноз нервно-мышечных заболеваний должен включать несколько основных характеристик: локализацию ведущего двигательного дефекта; тип наследования: аутосомно- доминантный, аутосомно-рецессивный, сцепленный с Х-хромосомой; возраст при появлении первых признаков заболевания; преимущественную локализацию поражения скелетных мышц; течение заболевания (быстро прогрессирующее, медленно прогрессирующее). В детской практике важное значение имеет диагностика синдрома «вялого ребенка». Клинические проявления синдрома включают:
Нео6ычную позу (поза «лягушки»);
Слабое сопротивление при пассивных движениях за счет мышечной гипотонии; ,
Увеличение объема движений в суставах;
Снижение общей двигательной активности;
Задержку моторного развития.
Данный симптомокомплекс характерен для широкого спектра заболеваний нервно-мышечной системы, перинатальных поражений ЦНС, заболеваний соединительной ткани. Кроме того, данный симптомокомплекс может проявляться при тяжелой соматической патологи новорожденного.
Врожденные миопатии - группа наследственных заболеваний мышечной ткани. Эти заболевания проявляются низким мышечным тонусом, слабостью мышечной мускулатуры, снижением сухожильных рефлексов. Часто наблюдаются дыхательные нарушения, приводящие к пневмониям.
Выделяют несколько форм заболевания. Важное значение для диагностики имеет электромиографическое исследование.
Наибольшее значение в период новорожденности имеет диагностика особой формы мышечной дистрофии, которая сочетается с ограничением движений глазных яблок. Уже с рождения характерны низкий мышечный тонус, ограничение движений глазных яблок. Ребенок плохо сосет; у него слабый, тихий крик; наблюдаются признаки бульбарного паралича. Заболевание медленно прогрессирует, все более отчетливо проявляется бульбарная дизартрия.
Врожденная непрогресирующая или медленно прогрессирующая немалиновая миопатия - Заболевание наиболее часто проявляется в дошкольном возрасте в виде жалоб на мышечную утомляемость, слабость, задержку психо-речевого развития. Этим детям нередко ошибочно ставят диагноз ДЦП. При осмотре такого ребенка обращают на себя внимание диффузные мышечные гипотрофии, быстрая утомляемость, мышечная слабость, снижение сухожильных рефлексов. Существует несколько форм миопатий.
Врожденные мышечные дистрофии - это наследственные заболевания с аутосомно-рецессивным типом наследования.
Наиболее распространенными являются прогрессирующие мышечные дистрофии. В раннем возрасте наиболее часто наблюдается форма Дюшенна.
Миопатия Дюшенна (1868г.). Среди всех мышечных дистрофий псевдогипертрофическая форма Дюшенна самая тяжелая и самая распространенная. Она отмечается в популяции с частотой 1:30 000.
Клиническая картина - прогрессирующее нарастание мышечных дистрофий, дистрофических изменений с постепенным обездвиживанием. Поражаются прежде всего нижние конечности. Первые симптомы появляются в возрасте двух-четырех лет, хотя уже на первом-втором году жизни двигательная активность больных снижена. Дети позднее начинают ходить, не бегают, развивается специфическая «утиная» походка с переваливанием. Процесс восходящий - ноги, мышца спины, грудной клетки, верхних конечностей. К 13-15 годам полностью обездвижены. Смерть наступает в конце второго десятилетия, чаще от острой сердечной недостаточности или пневмонии. 30-70% больных миопатией Дюшенна имеют умственную отсталость. Степень интеллектуального недоразвития различная. Может сопровождаться отставанием в формировании речи, вызывается рецессивным геном, локализованным в Х-хромосоме, предполагается, что это нарушение связано с дефицитом мембран мышечного волокна и другими причинами.
Наследственные нервно-мышечные болезни поддаются лечению с помощью генной терапии. В 1985 г. был открыт дистрофиновый ген, дефект которого приводит к миотонической дистрофии Дюшенна. Больным детям вводят мышечные клетки (миобласты), полученные из биопсий (кусочек мышцы весом около 1-2 г) здоровых доноров и выращенные при определенных условиях. Эти здоровые миобласты несут в себе отсутствующий дистрофиновый ген. В мышцах больного донорские клетки сливаются с клетками пациента, и формируются мышечные волокна с полным набором генов.
Родителями больных детей был создан Московский фонд помощи детям, больным нервно-мышечными болезнями и общая межрегиональная ассоциация «Надежда». Главная их цель - поддержка семей, имеющих детей с миодистрофией Дюшенна, и организация лечения.
Миотоническая дистрофия (атрофическая миотония, дистрофическая миотония, болезнь Россолимо-Куршманна - Стейнерта - Баттена) частота 1 на 8000. Мужчины болеют чаще в 3 раза. Ген локализован на 19q 13.2-13.3. Наследуется по аутосомно-доминантному типу. Основные симптомы: слабость в руках, раннее облысение, гипогонадизм, патология сердца, эндокринные нарушения, интеллектуальное снижение, почти всегда ограничение подвижности глазных яблок и птоз.
Среди всех случаев миотопической дистрофии врожденная форма - болезнь Томпсена - 12%. Синоним - миотопия врожденная - резко выраженный двусторонний парез лицевой мускулатуры - лицевая диплегия: лицо амимично, рот открыт, больной не может наморщить лоб, зажмурить глаза, улыбнуться. Встречаются врожденные уродства: косолапость, гиперостозы и асимметрия черепа, артрогрипозы. Нёбо всегда резко сужено, высокое. Имеют очень тонкие рёбра. Нередко затруднение глотания, часто бывают пневмонии, сердечные аритмии. Отмечается своеобразный рост волос на лбу - «залысины» с обеих сторон. Умственная отсталость встречается у всех пораженных IQ от 20-70, катаракта.
Заболевание вызывается аутосомно-доминантным геном с резко варьирующейся экспрессивностью.
По некоторым данным, для женщины, страдающей миотонической дистрофией, риск иметь ребенка с тяжелой конгенитальной формой болезни составляет около 7% , а если уже был один ребенок с таким поражением, то 35%. В настоящее время делаются попытки пренатальной диагностики носительства гена миотонической дистрофии на основании анализа сцепления этого гена с локусом секретора и группой крови.
Важное значение имеет ранняя диагностика спинальных мышечных атрофий у детей раннего возраста (подавляющее большинство этих атрофий наследуется по аутосомно-рецессивному типу).
Спинальные мышечные атрофии детского возраста. Наследуются по аутосомно-рецессивному типу одной из таких форм является Верднига-Гоффмана. Ген локализован на 5q 11.2-13.3. Во время беременности отмечается позднее, вялое шевеление плода. С рождения присутствует генерализованная мышечная гипотония, уже в первые месяцы возникают атрофии и подергивания мышц спины, туловища, верхних и нижних конечностей. Рано наблюдается отставание моторного развития.
Дегенеративные заболевания с преимущественным поражением нервно-мышечного аппарата составляют самую значительную группу среди всех наследственных заболеваний.
Исключительно важными, а нередко решающими в диагностике нервно-мышечных заболеваний являются результаты электрофизиологических и биохимических исследований. Столь же велико значение патоморфологических находок. Изучение мышечного биоптата в световом микроскопе помогает дифференцировать миогенную атрофию от неврогенной. Гистохимическое исследование необходимо для выявления метаболических поражений мышц, а электронная микроскопия открыла целый большой класс заболеваний - непрогрессирующие миопатии.
Прогрессирующие мышечные дистрофии. Термином мышечные дистрофии называется группа генетически детерминированных расстройств, характеризующихся прогрессирующими дегенеративными изменениями в мышечных волокнах без первичной патологии периферического (нижнего) мотонейрона.
Различные формы отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных атрофии, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.
Большинство мышечных дистрофий хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросы их патогенеза и лечения остаются до сего времени неразрешенными. Большие надежды возлагаются на молекулярную генетику, с помощью которой определено местонахождение генов уже многих нозологических форм.
Диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число членов семьи затрудняет определение типа наследования.
Характерным моторным дефектом у больных с мышечными дистрофиями является "утиная" походка: больной ходит переваливаясь к боку на бок. Она связана главным образом со слабостью ягодичных мышц, прежде всего средней и малой, которые фиксируют таз относительно бедренной кости. В результате при заболевании возникает наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). При ходьбе сторона наклона постоянно меняется. Указанные изменения можно проверить и в пробе Тренделенбурга, попросив больного поднять одну ногу, согнув ее под прямым углом в коленном и тазобедренном суставах: таз на стороне поднятой ноги опускается (а не поднимается как в норме) из-за слабости средней ягодичной мышцы опорной ноги.
Поднимаясь из горизонтального положения, больной с выраженной мышечной слабостью проксимальных мышц с трудом переворачивается на живот, затем, упираясь руками в пол, становится на четвереньки и после этого, упираясь руками в голени, затем в бедра, постепенно выпрямляется. Этот феномен "набирания по себе" носит название приема Говерса. Часто он связан со слабостью больших ягодичных мышц.
Миодистрофия Дюшенна. Псевдогипертрофическая мышечная дистрофия Дюшенна встречается чаще всех других заболеваний мышечной системы (30 на 100000 живых новорожденных). Характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У детей более раннего возраста начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать. Больные умирают на 2-3-м десятилетии жизни.
Одними из первых признаков заболевания являются уплотнения икроножных мышц и постепенное увеличение их объема за счет псевдогипертрофий. Атрофии мышц бедра, тазового пояса нередко маскируются хорошо развитой подкожной жировой клетчаткой. Постепенно процесс принимает восходящее направление и распространяется за плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук.
В терминальной стадии слабость мышц может распространяться на мышцы лица, глотки, дыхательные мышцы.
В развитой стадии болезни имеются такие характерные симптомы, как "утиная походка"; подчеркнутый поясничный лордоз, крыловидные лопатки, симптом "свободных надплечий". Типичны ранние мышечные контрактуры и сухожильные ретракции, особенно ахилловых сухожилий. Рано выпадают коленные рефлексы, а затем рефлексы с верхних конечностей.
Псевдогипертрофии могут развиваться не только в икроножных, но также в ягодичных, дельтовидных мышцах, мышцах живота, языка. Очень часто страдает сердечная мышца по типу кардиомиопатии. Выявляются нарушения ритма сердечной деятельности, расширение границ сердца, глухость тонов, изменения ЭКГ. Острая сердечная недостаточность - наиболее частая причина летальных исходов при миодистрофии Дюшенна. На вскрытии находят фиброз и жировую инфильтрацию сердечной мышцы.
Нередко наблюдается нарушение моторики желудочно-кишечного тракта.
Частым симптомом является снижение интеллекта. Представляет интерес тот факт, что в одних семьях олигофрения бывает резко выражена, в других сравнительно умеренно. Изменение высших психических функций обычно не прогрессирует и не коррелирует с тяжестью мышечного дефекта. Оно не может быть объяснено только педагогической запущенностью больных детей, которые рано выключаются из детских коллективов, не посещают детский сад и школу из-за двигательных дефектов. При КТ и МРТ нередко обнаруживают церебральную атрофию, возможно, связанную с нарушением пренатального развития головного мозга.
Нередко у детей развивается адипозогенитальный синдром, иногда и другие признаки эндокринной недостаточности. Часто находят изменения в костной системе: деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз.
Отличительной особенностью формы Дюшенна является высокая степень гиперферментемии уже на ранних стадиях развития процесса. Так, уровень специфического для мышечной ткани фермента - креатининфосфокиназы - в сыворотке крови может превышать в десятки и даже сотни раз нормальные показатели. Резкое (в 10-100 раз) увеличение креатининфосфокиназы (КФК) при нервно-мышечной патологии должно побуждать к обсуждению прежде всего следующих заболеваний: болезни Дюшенна, болезни Беккера, полиомиозита и дерматомиозита, пароксизмалыюй миоглобулинурии, дистальной миодистрофии. Лишь в далеко зашедших стадиях болезни степень гиперферментемии постепенно снижается. Имеются сообщения о повышении КФК на стадии внутриутробного развития.
Миодистрофия Дюшенна передается по рецессивному, сцепленному с Х-хромосомой типу. Ген локализован в коротком плече Х-хромосомы. Довольно высока частота мутации гена (30%), что объясняет большое количество спорадических случаев.
Мутация (чаще всего делеция) приводит к половому или почти полному отсутствию продукта гена - структурного белка дистрофика. Физиологическая роль дистрофика до конца не установлена. Он обнаруживается в больших концентрациях в области сарколеммы, играя, видимо, определенную роль в поддержании целости этой мембраны. Отсутствие дистрофика вызывает структурные изменения в сарколемме, что в свою очередь приводит к потере внутриклеточных компонентов и повышенному входу кальция, что в конечном счете ведет к гибели миофибрилл. Полагают, что дефицит дистрофика в синаптических зонах корковых нейронов является причиной задержки психического развития.
Для медико-генетического консультирования очень важно установление гетерозиготного носительства. При миодистрофии Дюшенна у гетерозигот приблизительно в 70% случаев выявляются субклинические, а иногда и явные признаки мышечной патологии - некоторое уплотнение и даже увеличение икроножных мышц, быстрая утомляемость мышц при физической нагрузке, изменения на ЭМГ и при патоморфологическом исследовании мышечных биоптатов. Наиболее часто у гетерозиготных носительниц выявляется увеличение активности креатининфосфокиназы.
При наличии клинической картины миодистрофии Дюшенна у лиц женского пола следует в первую очередь исключить возможность аномалии по Х-хромосоме - синдром Шерешевского-Тернера (ХО), синдром Морриса (XY) или мозаицизм по этим синдромам.
Мышечная дистрофия Дюшенна, начинающая развиваться еще в пренатальном периоде, является по сути врожденной миопатией и может быть диагностирована вскоре после рождения путем проведения мышечной биопсии и определения активности КФК.
Миодистрофия Беккера. Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофией Дюшенна существует доброкачественная форма - болезнь Беккера. По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягко, больные длительно сохраняют работоспособность, в возрасте 20- 30 лет и позже еще могут ходить. Фертильность не снижена, поэтому заболевание иногда прослеживается в нескольких поколениях семьи: больной мужчина через свою дочь передает заболевание внуку ("эффект деда"). Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракция пяточных (ахилловых) сухожилий выражена менее резко, чем при болезни Дюшенна.
При этой форме не отмечается нарушений интеллекта, кардиомиопатия отсутствует или выражена незначительно.
Как и при других Х-сцепленных миодистрофиях, при форме Беккера значительно повышается активность КФК, хотя и в меньшей степени, чем при болезни Дюшенна, не превышая 5000 ед. Ген болезни Беккера, как и болезни Дюшенна, локализуется в коротком плече Х-хромосомы; вероятно, оба локуса тесно связаны между собой или являются аллельными. В отличие от болезни Дюшенна, при которой дистрофии практически отсутствует, при болезни Беккера синтезируется аномальный дистрофии. Отличия обнаруживаются и при мышечной биопсии. При миодистрофии Беккера мышечные волокна обычно неокруглы, гиалиновые волокна, характерные для миодистрофии Дюшенна, наблюдаются крайне редко.
Миодистрофия Ландузи-Дежерина (лицелопаточно-плечевая миодистрофия). Заболевание передается по аутосомно-доминантному типу с высокой пенетрантностью, но несколько вариабельной экспрессивностью. Встречается гораздо реже, чем миодистрофия Дюшенна (0,4 на 100 тыс. населения). Предполагают, что ген этого заболевания локализован в 4-й хромосоме. Женщины болеют чаще мужчин (3:1), Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.
Миодистрофия Ландузи-Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она еще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике, удается выявить некоторую слабость мышц, например мышц лица, и в более раннем возрасте.
Мышечная слабость и атрофия вначале появляются в мышцах лица или плечевого пояса. Постепенно эти нарушения распространяются на мышцы проксимальных отделов рук, а затем и на нижние конечности. В большинстве случаев вначале поражаются мышцы передней поверхности голеней (с развитием свисающей стопы), затем мышцы проксимальных отделов ног. На высоте заболевания грубо страдают круговые мышцы глаза и рта, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая, трехглавая мышцы плеча. Характерен внешний вид больных: типичное лицо миопата с "поперечной улыбкой" ("улыбка Джоконды"), протрузией верхней губы ("губы тапира"), резко выраженные крыловидные лопатки, своеобразная деформация грудной клетки с уплощением ее в переднезаднем направлении и ротацией внутрь плечевых суставов. Нередко имеется асимметрия поражения, даже в пределах одной мышцы (например, круговой мышцы рта). Может наблюдаться псевдогипертрофия икроножных, дельтовидных мышц, иногда мышц лица. Контрактуры и ретракции выражены умеренно. Сухожильные рефлексы длительное время бывают сохранены, но иногда снижаются уже на ранней стадии.
Признаки поражения сердечной мышцы выявляются редко. Активность сывороточных ферментов увеличена незначительно и может быть нормальной. Интеллект не страдает. Продолжительность жизни в большинстве случаев не снижается. Представляет интерес тот факт, что ЭМГ при миодистрофии Ландузи-Дежерина нередко не совсем типична для мышечного уровня поражения. У некоторых больных (членов одной семьи) может наблюдаться снижение амплитуды биопотенциалов, интерференционный тип кривой, у других, наоборот, уменьшение частоты и гиперсинхронная активность, иногда с типичным ритмом частокола. Следует помнить о спинальном варианте, имитирующем болезнь Ландузи-Дежерина.
Миодистрофия Эрба-Рота (конечностно-поясная миодистрофия). Передается по аутосомно-рецессивному типу, оба пола страдают одинаково. Начало заболевания в большинстве случаев относится к середине 2-го десятилетия жизни (14-16 лет), однако описана как ранняя, псевдодюшенновская форма, когда первые симптомы проявляются в возрасте до 10 лет и заболевание протекает тяжело, так и поздний вариант с началом после 30 лет.
Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидизация наступает через 15-20 лет от начала появления первых симптомов. Миодистрофия начинается либо с поражения мышц тазового пояса и проксимальных отделов ног (форма Лейдена-Мебиуса), либо с плечевого пояса (форма Эрба). В некоторых случаях плечевой и тазовый пояса поражаются одновременно. Довольно значительно страдают мышцы спины и живота. У больных имеется характерная "утиная" походка, затруднено вставание из положения лежа и сидя, подчеркнут поясничный лордоз. Мышцы лица в большинт стве случаев не страдают. Для этой формы малохарактерны контрактуры и псевдогипертрофии. Могут иметь место концевые атрофии и сухожильные ретракции. Интеллект обычно сохранен. Сердечная мышца большей частью не поражена. Уровень ферментов в сыворотке крови, как правило, повышен, однако не столь резко, как при Х-сцепленной миодистрофии. Есть указания, что у больных мужского пола уровень КФК выше, чем у больных женщин. Имеется значительная разница в экспрессивности мутантного гена у разных членов семьи - наряду с тяжелой клинической картиной могут быть сравнительно легкие и даже стертые клинические симптомы. Смерть обычно наступает от легочных осложнений.
Поскольку клинику конечностно-поясной миодистрофии особенно охотно имитируют нервно-мышечные заболевания иного характера, необходимо, особенно в спорадических случаях и при позднем начале заболевания, проводить тщательное клиническое обследование для исключения спинальной амиотрофии, полимиозита, метаболических, эндокринных, токсических, лекарственных, карциноматозных миопатий. В прошлом имела место явная гипердиагностика этой формы мышечных дистрофий.
Лечение мышечных дистрофий. Терапевтические возможности при мышечных дистрофиях весьма ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и, возможно, на некоторое снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Лечебный комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию.
Лечебная гимнастика состоит из пассивных и активных движений, выполняемых во всех суставах в различных положениях: стоя, сидя, лежа, при различном положении конечностей. Активные движения предпочтительнее выполнять в изометрическом режиме. Занятия гимнастикой необходимо проводить регулярно по нескольку раз в день. В то же время следует предостеречь от чрезмерных упражнений, особенно сопровождающихся перерастяжением мышц. Важное значение (особенно после иммобилизации больного) имеют дыхательные упражнения.
Ортопедические мероприятия консервативного (специальные шины) и оперативного характера (ахиллотомия, пересечение икроножной мышцы), направленные на коррекцию контрактур и формирующихся патологических установок конечностей, также имеют целью сохранить возможность самостоятельного передвижения. При этом в каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. Следует учитывать, что нередко (в частности, при выраженном гиперлордозе и слабости четырехглавой мышцы бедра) эквиноварусная установка стоп имеет компенсаторное значение и после проведения, например, ахиллотомии больной может оказаться окончательно обездвиженным. При развивающихся контрактурах рекомендуется проводить осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Медикаментозная терапия предполагает назначение препаратов метаболического действия, направленных на восполнение энергетического и белкового дефицита, однако их эффективность весьма сомнительна. Применяют антагонисты кальция (в связи с выявленным при болезни Дюшенна дефектом клеточных мембран, приводящим к повышенному поступлению кальция внутрь клетки), иммуномодуляторы, фосфорсодержащие соединения (АТФ, фосфаден), витамин Е (100мг внутрь 3 раза в день). Показано, что при болезни Дюшенна применение преднизолона (0,75 мг/кг в сутки) может драматически увеличивать силу мышц, однако этот эффект сохраняется неболее года и в целом не влияет на исход заболевания. В связи с серьезными побочными эффектами, возникающими ври длительном применении препарата, его использование нецелесообразно. Оценки эффекта анаболических стероидов противоречивы и их назначение зачастую сопряжено с неоправданным риском. Оценивая эффект тех или иных препаратов при болезни Дюшенна, следует учитывать, что при умеренной выраженности заболевания у больных в возрасте 3-6 лет может отмечаться относительная стабилизация состояния, связанная с возрастным развитием мышечной системы, приобретением двигательных навыков, что может в какой-то степени временно скомпенсировать непрерывно текущий дистрофический процесс.
Определенное значение имеет коррекция питания больного, рекомендуется диета с высоким содержанием белка и низким содержанием жиров и пониженной калорийностью при оптимальном содержании витаминов и микроэлементов. Важную роль играет психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
В настоящее время принято считать, что термин «миопатия» следует использовать для обозначения всех заболеваний скелетных мышц. Термин «мышечная дистрофия» применяют к наследственным формам первичных миопатий с прогрессирующим течением.
Мышечные атрофии, обусловленные неврогенным дефектом, считают вторичными и называют «амиотрофиями» спинальными или невральными.
Некоторые авторы, особенно в отечественной литературе, пользуются термином «миопатия» применительно к первичным поражениям мышц как синонимом прогрессирующей мышечной дистрофии, что не совсем точно отражает характер процесса.
Миопатией можно называть группу непрогрессирующих мышечных поражений, связанных или с морфологическим дефектом (например, митохондриальные миопатии), или с нарушением метаболизма (гликогеновые миопатии и т. д.)
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
При гипокалиемической пароксизмальной миоплегии в момент приступов отмечается снижение уровня сывороточного калия до 2 ммоль/л и даже ниже. Это не сопровождается усилением экскреции иона калия с мочой, в связи с чем предполагается, что в момент приступов гипокалиемической миоплегии происходит перемещение этих ионов внутрь клеток. Исходя из представлений о таком перемещении ионов калия внутрь мышечных волокон,…
Достоверное повышение концентрации натрия отмечено и в эритроцитах больных гипокалиемической миоплегией [Ильина Н. А. и др., 1977]. Выраженный клинический эффект, наблюдавшийся у этих больных в период лечения диакарбом, сопровождался нормализацией содержания в эритроцитах натрия. В литературе, посвященной миоплегии, важная роль в механизме развития гипокалиемической ее формы отводится инсулину. Широко известна способность экзогенного инсулина провоцировать миоплегические…
Для выяснения возможной общности механизма деполяризации сарколеммы при разных формах периодического паралича целесообразно рассмотреть вопрос, какие изменения концентрации ионов или проницаемости для них могут обусловить данный феномен. Известно, что мембранный потенциал покоя зависит от концентрации натрия, калия и хлоридов по обе стороны мембраны и от относительной проницаемости мембраны для этих ионов. В нормальных условиях в…
Лечение пароксизмальной миоплегии проводится строго дифференцированно в соответствии с формой первичной миоплегии или характером основного заболевания при фенокопиях миоплегии. В лечении семейного гипокалиемической формы паралича важное значение придается диете. Рекомендуется ограничение общей калорийности суточного рациона и особенно избегать неумеренного приема углеводов. Целесообразно также ограничить прием поваренной соли. В ряде случаев выполнение этих диетических рекомендаций оказывает…
С 1960 г. для профилактики приступов гипокалиемической формы миоплегии успешно применяется верошпирон по 100 - 300 мг ежедневно. В большинстве случаев данная терапия приводит к значительному уменьшению частоты и выраженности приступов [Ильина Н. А., 1973; P. Cassa, 1964]. Однако у некоторых больных верошпирон не оказывает терапевтического действия [Аверьянов Ю. Н., 1977]. Известно также, что верошпирон…
При лечении больных гиперкалиемической пароксизмальной миоплегией нужно учитывать, что продукты с высоким содержанием калия способствуют развитию приступов. Рекомендуется включать в рацион достаточное количество углеводов, а также несколько повышенное количество поваренной соли. Рекомендуется дробный прием пищи с укороченными интервалами между отдельными приемами. Выраженное купирующее влияние на уже развившийся приступ гиперкалиемической миоплегии оказывает внутривенное введение глюкозы с…
Гипокалиемическая пароксизмальная миоплегия является наиболее часто встречающейся формой миоплегии. Все исследователи отмечают значительное преобладание среди больных с данной формой периодического паралича лиц мужского пола. Helveg-Larsen Н. и соавт. (1955) сообщили, что из обследованных ими 34 больных мужчин было 31, а женщин всего 3. Из 55 больных гипокалиемической пароксизмальной миоплегией, наблюдавшихся нами, мужчин было 44, женщин…

Клиническая картина гиперкалиемической формы миоплегии (эпизодическая адинамия) была наиболее тщательно изучена J. Gamstorp (1956, 1957). Заболевание наследуется аутосомно-доминантно с полной пенетрантностью и одинаково часто встречается у лиц того и другого пола. Первые приступы появляются, как правило, в первом десятилетии жизни, причем у преобладающей части больных в первые 5 лет. Родословная семьи С-вой Гиперкалиемическая форма пароксизмальной…
В 1961 г. D. Poskanzer и D. Kerr представили описание третьей формы семейной миоплегии - нормокалиемической. В исследованной ими семье у 21 человека отмечались приступы миоплегии, не сопровождавшиеся изменениями содержания сывороточного калия. Тип наследования в данной семье аутосомно-доминантный с полной пенетрантностью. У большинства больных заболевание началось в первом десятилетии жизни. Приступы варьировали в выраженности, в…
В ряде случаев приступы миоплегии возникают как осложнения того или иного заболевания или медикаментозной терапии и тогда рассмотриваются как фенокопии наследственного заболевания. Наиболее частой причиной вторичной формы миоплегии является тиреотоксикоз. Имеются указания на значительное преобладание среди больных тиреотоксической пароксизмальной миоплегией японцев, китайцев и корейцев. По данным A. Engel (1961), из 228 описанных на то время…






